How to perform de novo assembly with PATRIC
De novo assembly is a method for generating contigs, scaffolds, or complete genomes from Next Generation sequencing reads (i.e. DNA fragments). Some researchers use the free, online bioinformatics tool PATRIC for de novo assembly. PATRIC includes several de novo assembly algorithms that are especially useful for microbial scale genomes. Although PATRIC includes tutorials on how to use its de novo assembly tools, we’ll provide here a more condensed version that some researchers find easier to follow.
Create a PATRIC account
We’ll assume that you have already created your own PATRIC account (see “Register” on the PATRIC home page), logged into the account, and created your own workspace directories. These activities are beyond the scope of this brief article. And we’ll assume that you have already completed a sequencing run, retrieved your project FastQ files, and stored those files somewhere on your local workstation.
Required Files
Furthermore, we’ll assume that you have two Illumina, gzip-compressed, paired-end, FastQ read files with the following file names stored on your local workstation:
- test_assembly_R1.fastq.gz
- test_assembly_R2.fastq.gz
These files are typically generated from sequencing runs and we deliver them to clients via Box accounts or other means. Most bioinformatics post-processing analyses begin with FastQ files, including de novo assembly. Note that the files are differentiated by the “R1” (forward-read) and “R2” (reverse-read) designations.
How to perform a PATRIC de novo assembly run
Please follow these steps to perform a PATRIC de novo assembly run.

Fig. 1
Navigate to your home directory by choosing “WORKSPACES -> home” in the main menubar. Create a top-level directory to contain the read files and output directory by choosing “ADD FOLDER” in the upper right-hand corner. We’ll call this directory “Assembly” (Fig. 1). Navigate into the directory “Assembly” by clicking the folder icon to the left of the directory name.

Fig. 2
Once you’re in the “Assembly” directory, there should be an “ADD FOLDER” icon in the upper right-hand corner. Click “ADD FOLDER” and create a new subdirectory, which will eventually contain all of the output files from the assembly run. We’ll call the subdirectory “assembly_output” (Fig. 2).

Fig. 3
In the top menubar, click “SERVICES -> Genomics -> Assembly” (Fig. 3). This should display a “Genome Assembly” page.
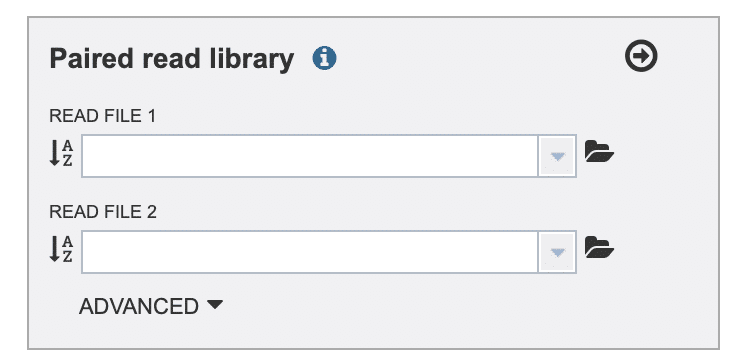
Fig. 4
In the “Paired read library” selection box (Fig. 4), click the folder icon to the right of the “READ FILE 1” drop-down list. This should display a “Choose or Upload a Workspace Object” page.
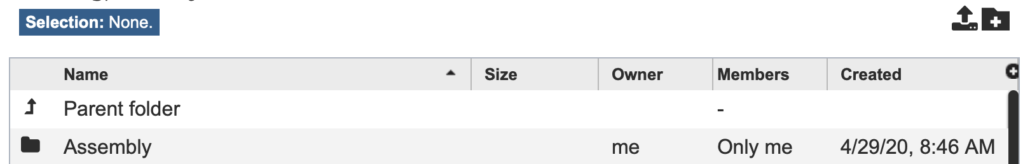
Fig. 5
On this page, you should see the “Assembly” top-level directory and a folder icon next to it (Fig. 5). Click the folder icon to enter the “Assembly” directory.
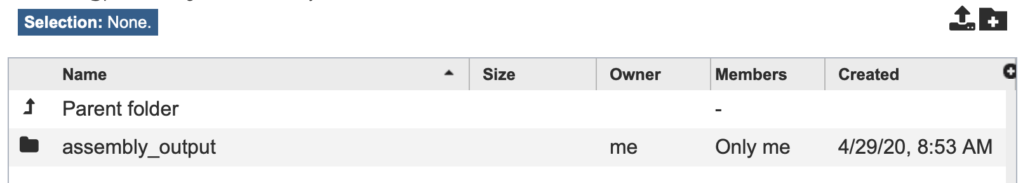
Fig. 6
In the upper right-hand corner of this page, there should be an arrow icon (Fig. 6). If you hover the mouse pointer over this icon it should say “Upload to current folder”. Click the arrow icon. This should display an “Upload files to Workspace” page.
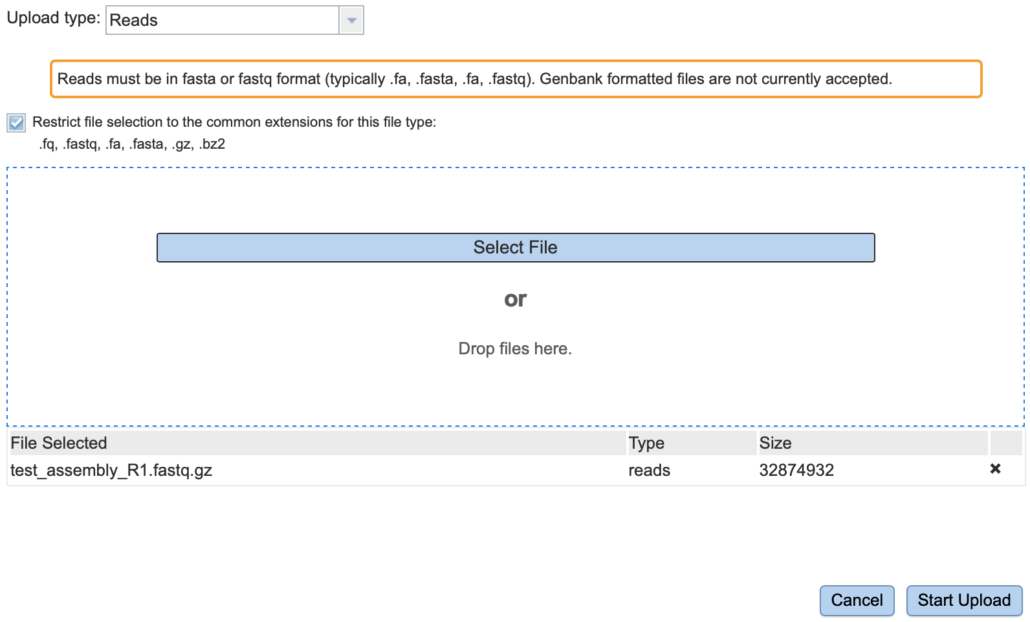
Fig. 7
On this page, in the “Upload type:” drop-down list, choose “Reads” (Fig. 7). Check the box “Restrict file selection to the common extensions for this file type:”. Click “Select File”. At this point, you should be able to navigate to the file “test_assembly_R1.fastq.gz” on your local workstation. Make sure you choose the file with “R1” in the file name. The file name should appear under the “File Selected” header. Choose “Start Upload” to upload the file. You should now see the contents of the “Assembly” directory.

Fig. 8
In the lower right-hand corner of this page, observe the progress percentage counter (Fig. 8). Wait for the counter to reach 100% before proceeding.
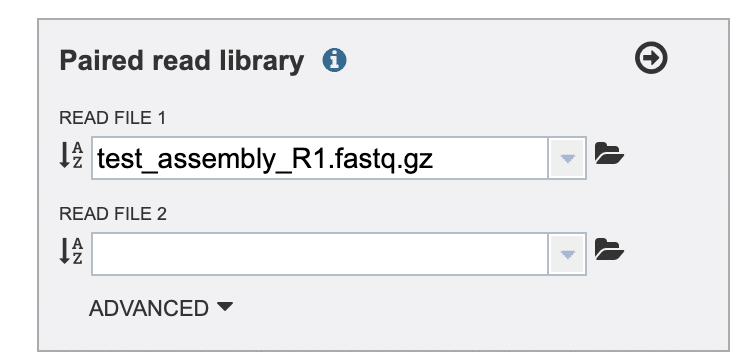
Fig. 9
You should now see the “Paired read library” selection box with the “test_assembly_R1.fastq.gz” file name in the “READ FILE 1” drop-down list (Fig. 9).
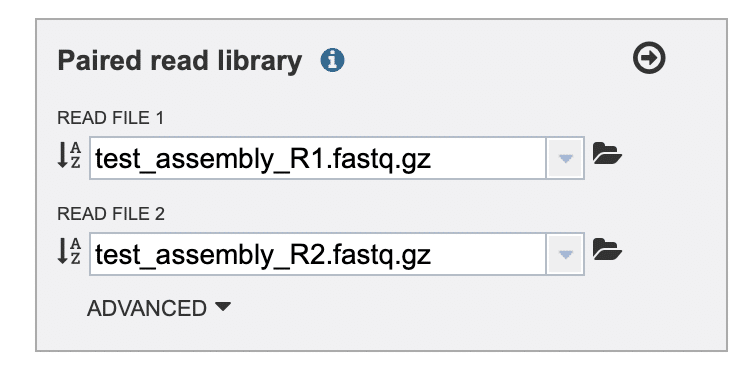
Fig. 10
Repeat these steps to upload file “test_assembly_R2.fastq.gz” into the “READ FILE 2” drop-down list. You should now see both read files displayed in the “Paired read library” selection box (Fig. 10). In the upper right-hand corner of the selection box, click the right arrow.
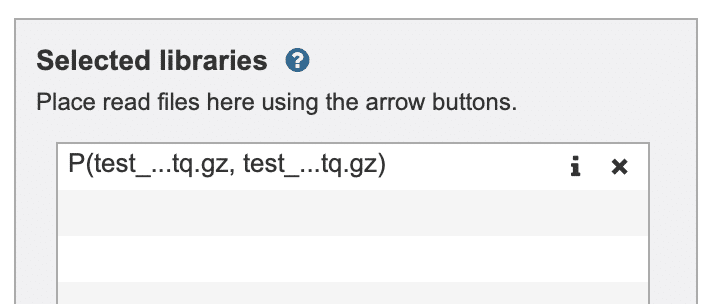
Fig. 11
This step will place the paired-end read files in the “Selected libraries” box (Fig. 11).
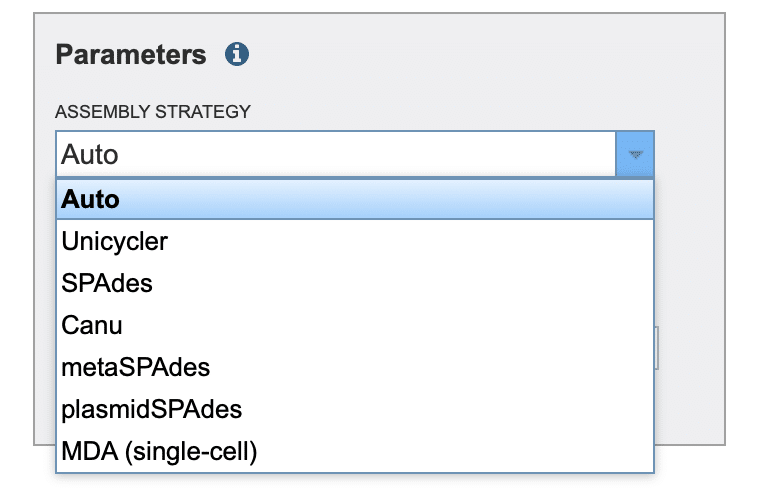
Fig. 12
In the “Parameters” selection box, click the “ASSEMBLY STRATEGY” drop-down list and choose one of the de novo assembler algorithms (Fig. 12). We’ll choose “SPAdes” for this example run.
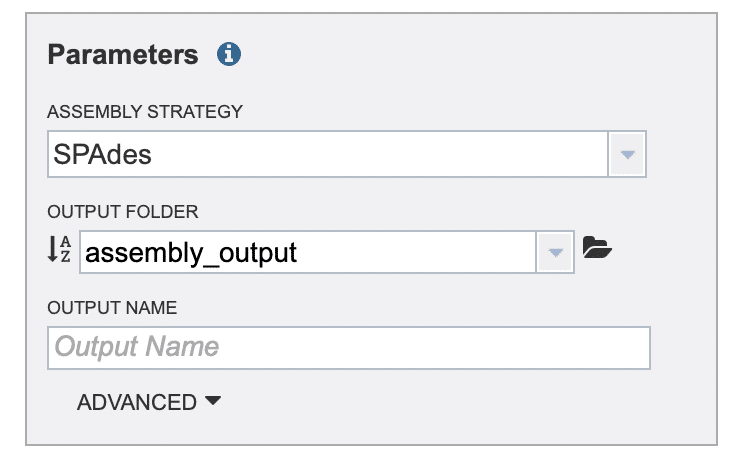
Fig. 13
In the “OUTPUT FOLDER” drop-down list, click the folder icon to the right and navigate to the “assembly_output” directory that we created earlier (Fig. 13).
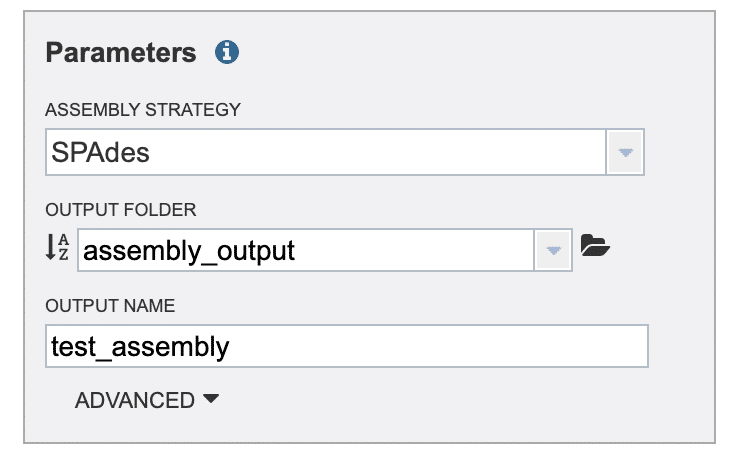
Fig. 14
In the “OUTPUT NAME” field, enter a descriptive name for the de novo assembly job (Fig. 14). Click the “Assemble” button to start the execution of the assembly run.
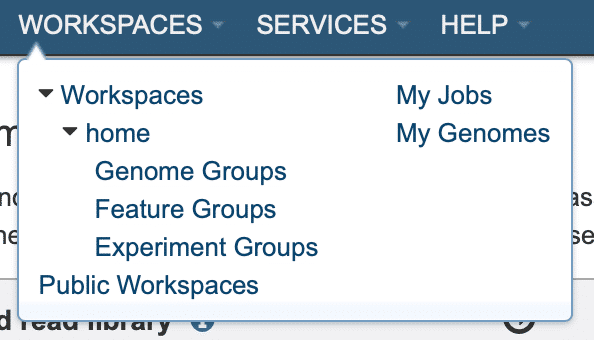
Fig. 15
To follow the status of your de novo assembly jobs, in the top menubar choose “WORKSPACES -> My Jobs” (Fig. 15).
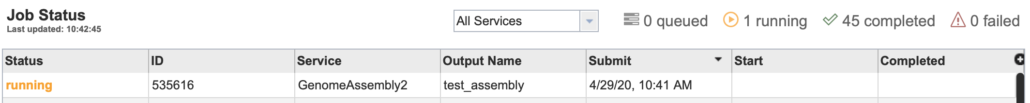

Fig. 16
This step will display a “Job Status” page including metadata about your queued, running, or completed jobs (Fig. 16).

Fig. 17
To view the results for a completed job, click the row of a specific job with “Status” = “completed”, which will highlight that row in blue (Fig. 17). Then choose “View” in the right-hand menubar. (You can click “Show” or “Hide” to view or not view job information).
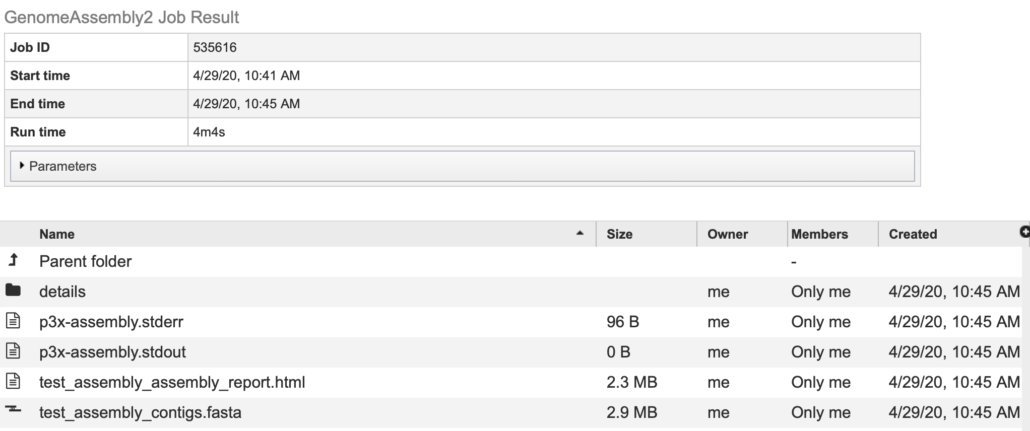
Fig. 18
You should see the contents of the “assembly_output” directory (Fig. 18). At this point, you can review the results of the de novo assembly job, download files or perform other post-processing activities.