What are paired-end reads?
Many sequencing library preparation kits include an option to generate so-called “paired-end reads“. In “short-read” sequencing, intact genomic DNA is sheared into several million short DNA fragments called “reads”. Individual reads can be paired together to create paired-end reads, which offers some benefits for downstream bioinformatics data analysis algorithms. The structure of a paired-end read is described here.
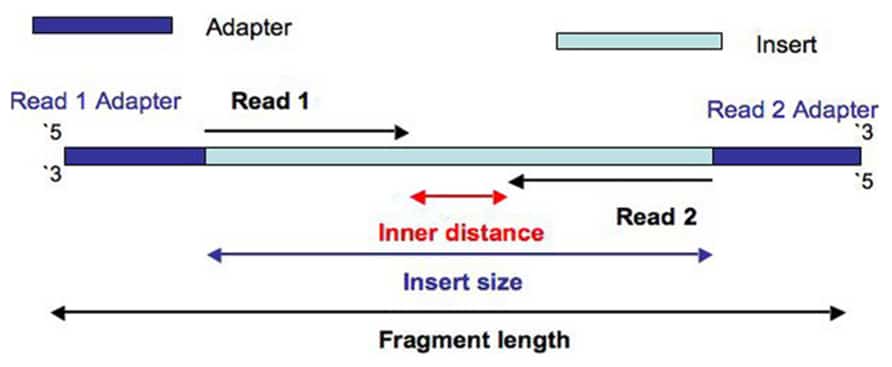
Fig. 1
Fig. 1 shows a schematic view of an Illumina paired-end read. There is a unique adapter sequence on both ends of the paired-end read, labeled “Read 1 Adapter” and “Read 2 Adapter”.
“Read 1”, often called the “forward read”, extends from the “Read 1 Adapter” in the 5′ – 3′ direction towards “Read 2” along the forward DNA strand.
“Read 2”, often called the “reverse read”, extends from the “Read 2 Adapter” in the 5′ – 3′ direction towards “Read 1” along the reverse DNA strand.
There is an arbitrary DNA sequence inserted between “Read 1” and “Read 2”, which we’ll call the “Inner sequence”. The length of this sequence is measured as the “Inner distance”. By definition, the “Insert” is the concatenation of “Read 1”, the “Inner distance” sequence and “Read 2”. And the length of the “Insert” is the “Insert size”. A single “Fragment” includes the “Read 1 Adapter”, “Read 1”, “Inner sequence”, “Read 2” and “Read 2 Adapter”. And the length of this “Fragment” is just the “Fragment length”.
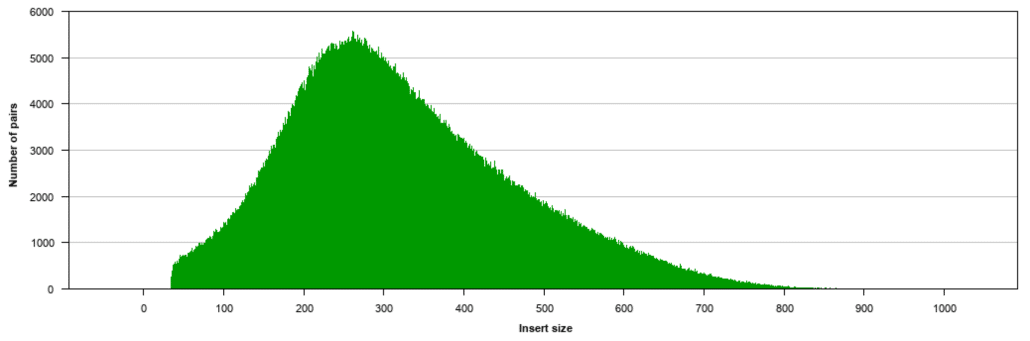
Fig. 2
Fig. 2 shows a typical insert size distribution for the Illumina Nextera XT DNA Library Preparation Kit. This is a probabilistic distribution and will vary somewhat for each DNA sample that is prepared with the XT kit. The distribution shows a peak insert size of around 300 bp. The distribution is somewhat leptokurtic and positively skewed with a minimum insert size around 40 bp and maximum insert size around 850 bp.
Note that due to the positively skewed nature of the distribution there is a significant number of paired-end reads with a fairly long total length (compared to just the individual reads themselves). This increase in total length is beneficial for sequence alignment algorithms, de novo assembly algorithms, spanning repetitive sequences, and the detection of insertions, deletions, and inversions.