How to generate a complete genome with hybrid assembly
Introduction
In this article, we’ll describe the Unicycler hybrid assembly pipeline. Unicycler is designed primarily for de novo assembly of bacterial genomes and includes the major steps required for hybrid assembly of sequenced microbial samples. It’s thus an instructive tool for understanding how to generate fully finished, complete genomes. The most current version of Unicycler is available on GitHub.
We’ll assume that we’re using Nanopore long-reads and Illumina short-reads for this discussion. There are other long-read (i.e. PacBio) and short-read (i.e. Qiagen) sequencing vendors but we’ll simplify things by restricting ourselves to Nanopore and Illumina.
Step 1: Long-read Assembly
Unicycler uses the miniasm de novo assembler and Racon consensus error correction tool for the assembly of Nanopore long-read sequences. Miniasm first attempts to find long-read overlap sequences with the minimap tool and with essentially no read error correction. It then builds a string graph representation of the nascent assembly. It then uses Racon to gap-fill the assembled reads into one or several consensus sequences.
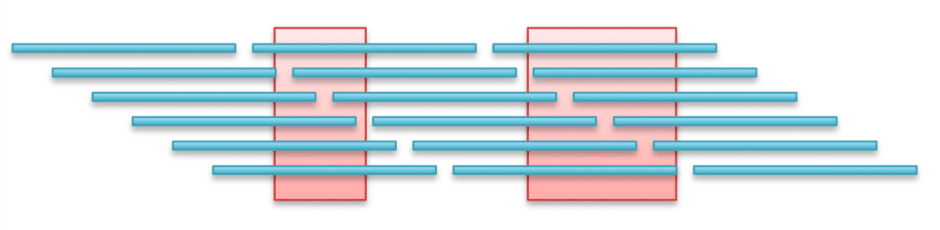
Fig. 1
Nanopore long-reads range in length from 500 bp to 2.3 Mb (Fig. 1; blue sequences). If there are repeat sequences in the sample genome (red sequences), long-reads can often span and exceed the entire length of the repeats. Furthermore, the base-calling accuracy for long-reads is now >95%.
Note that long-read assembly may or may not generate a fully finished, complete genome.
If the long-read assembly generates an acceptable finished genome, then we may be finished with the assembly pipeline and will not have to continue to Step 2. We can check the quality of the assembly with the methods shown in Step 5.
If the long-read assembly does not create a finished genome, or if the quality of the assembled genome is unacceptable, then we can move to Step 2 in the hybrid assembly pipeline and include high-quality Illumina short-read sequences.
Step 2: Short-read Assembly
Unicycler uses the SPAdes de novo assembler for the assembly of Illumina short-read sequences. There is an option to use Unicycler only for short-read assembly, but it’s deployed here as the second step in the hybrid assembly pipeline.
Before starting short-read assembly, Unicycler performs SPAdes read error correction to reduce the number of errors in subsequent assembly steps. This generally improves the performance of the assembly algorithms and also improves the final quality of assembled reads. SPAdes examines a range of k-mer sizes to maximize the chance of finding optimal assemblies for the final contigs and scaffolds.
As SPAdes assembles short-reads it scores an assembly graph for each k-mer to find an optimal k-mer size. SPAdes then performs various cleaning procedures on the assembly graph to remove overlaps and simplify the graph structure. SPAdes uses paired-end read information to help resolve short repeat sequences. This is one of several reasons we almost always use paired-end reads for short-read sequencing projects. Finally, SPAdes uses Pilon to improve the draft short-read assembly. Pilon can correct SNP’s, small indels, locally short misassemblies, and perform gap closure.
In almost all cases, short-read de novo assembly does not generate a fully finished, complete genome. Instead, it usually generates a few dozen to several hundred contigs and/or scaffolds. Short-read assemblies are inhibited from creating complete assemblies by the presence of repeat sequences and sometimes regions of high (>70%) or low (<30%) GC-content. Short-reads are simply unable to resolve these problematic genomic sequences. The assembly pipeline moves on to Step 3.
Step 3: Merge Assemblies
In this step, the hybrid assembly pipeline attempts to merge the Nanopore long-read and Illumina short-read assemblies into a single, finished genome. Unicycler takes advantage of the fact that long-reads can span repeat sequences and high or low GC-content regions, while the higher accuracy of short-reads (i.e. high Phred Q-scores) can be used to correct some of the lower quality long-read basepairs and perform some gap-filling actions.
Although the details of the hybrid assembly algorithm are beyond the scope of this article and are in fact fairly complex, we can understand the methodology at a high level. Using SPAdes, the hybrid assembler performs short-read assembly on the Illumina reads to generate high-quality contigs and scaffolds. It then attempts to create a string graph assembly from short-read contigs/scaffolds and long-reads. The string graph and long-reads are used to create bridges between sets of contigs/scaffolds, and then the bridge sequences themselves are merged into very long continuous sequences. At this point, Unicycler may have generated an almost complete genome. If there are small gaps remaining in the assembled genome, the pipeline moves to Step 4.
Step 4: Polish Assembly
In this step, Pilon and highly accurate short-reads are used to close any remaining small gaps, small indels, or mismatches in the assembly. Gap closure or gap-filling is used to generate a single, fully finished, complete genome.
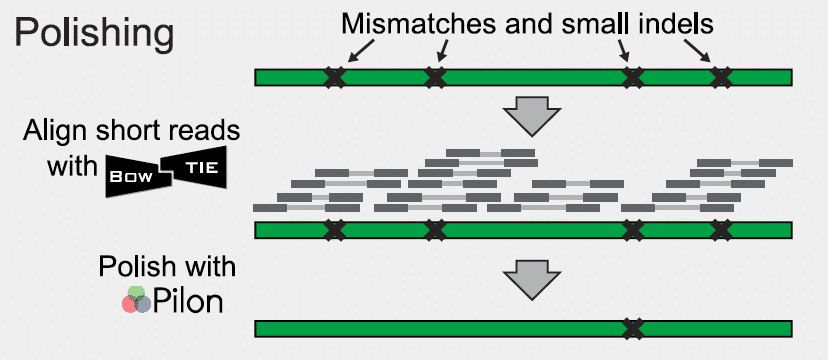
Fig. 2
When the assembly of contigs and scaffolds is complete, there are almost always gaps remaining in the sequence that must be closed or filled to finish the genome. As shown in Fig. 2, any remaining unmapped paired-end reads are mapped back to existing scaffolds. Some paired-end reads will overlap the remaining gaps between scaffolds. The overlapping reads are assembled locally at each gap location. If there is a sufficient number of overlapping reads at a particular gap, they will effectively close the gap. This process is repeated for all gaps in the sequence. If all gaps are closed in this fashion, the result will be a fully finished genome.
Step 5: Quality Control
There are several bioinformatics applications that you can use to assess the quality of de novo assembled genomes. Bandage displays assembly graphs, contigs, and the connections between contigs. It quickly and visually shows whether a de novo assembly algorithm generated a complete or fragmented genome. And it provides some metadata about the assembly that can be useful for further bioinformatics data analysis.

Fig. 3
For example, Fig. 3 shows the hybrid assembly of Helicobacter pylori into a single complete genome of length 1,646,095 basepairs. Note that the hybrid assembly also circularizes the bacterial genome as expected.

Fig. 4
In contrast, a short-read-only assembly of the same H. pylori sample yields a fragmented genome with 58 contigs (73 nodes in the assembly graph) as shown in Fig. 4. The distinction between hybrid assembly and short-read assembly is immediately apparent from these Bandage assembly graphs.
Assembly Inhibitors
Hybrid assembly is an excellent tool for building complete genomes. However, we must be aware that there are several issues that can lead to sequence inaccuracies, incomplete assemblies or misassemblies.
Among these are:
- Length of repeat sequences exceeds read length
- Multiple tandem repeats that collectively exceed read length
- High (>70%) or low (<30%) GC-content
- Assembly polishers “may” (rarely) introduce errors in repeat sequences
- Samples are not true isolates; sequence variations in mixed-species/strain samples
- Nearly identical plasmid sequences
- Long homopolymer sequences
- Methylation of nucleotide bases
- Inherent algorithmic issues (continuous improvements in assembler methodologies)
Unicycler does an excellent job of handling these issues and in most cases, they will not stop full genome assembly. Nevertheless, we should be cognizant that they can, in some cases, interfere with the assembly of fully finished genomes. In those cases, we can attempt to manually complete the genome or simply accept it as-is.
Example Assembly
Wadley et al. used Unicycler hybrid assembly with Nanopore long-reads and Illumina short-reads to generate a fully finished Escherichia coli chromosome and E. coli plasmid. The assembly was of sufficient quality for submission to the NCBI nucleotide database.
![]()
Fig. 5
Wadley et al. sequenced the ATCC type strain 11775 with a Nanopore MinION long-read instrument, yielding 118,118 reads with N50 = 15,397 bp (after performing quality scoring, filtering, and trimming). They obtained Illumina short-reads from an earlier project by Meier-Kolthoff et al., with NCBI deposition SRR3927310. They then performed Unicycler hybrid assembly with the combination of long-reads and short-reads. Fig. 5 shows Circos plots of the resulting complete circular genome (4,903,501 bp.) and complete circular plasmid (131,333 bp.). This is an excellent example of how we can create complete genomes and plasmids, down to the bacterial strain level, using hybrid de novo assembly of long-reads and short-reads.