Geneious sequence alignment & variant calls workflow
Geneious Prime is one of many bioinformatics tools that our clients use to post-process sequencing datasets. Geneious includes an extensive set of functions to perform sequence alignments of sample reads against reference genomes. And it includes functions to make variant calls and identify mutant alleles and small indels (insertions/deletions). We will briefly describe the Geneious sequence alignment and variant call workflow. We’ll assume the sample dataset was derived from an Illumina 2×150 bp paired-end read sequencing run.
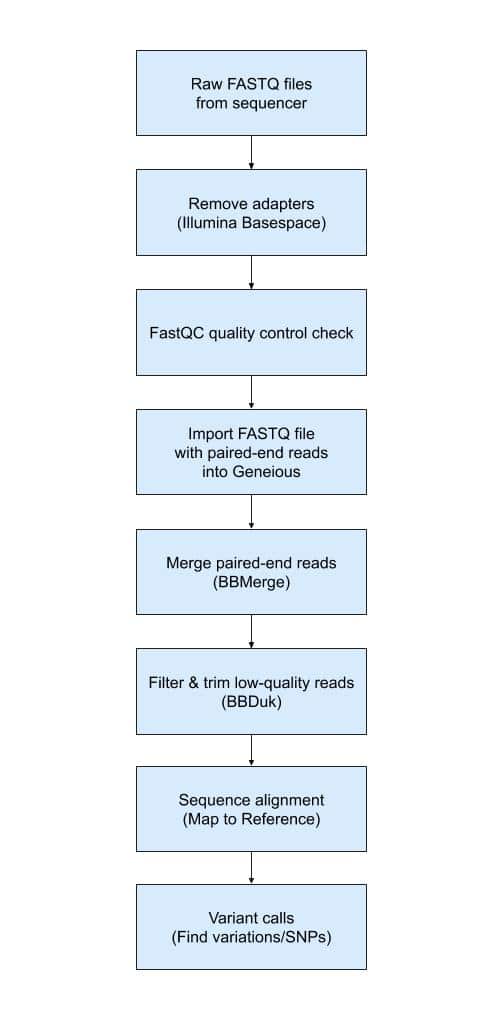
Fig. 1
Fig. 1 shows a high-level overview of the Geneious workflow. All bioinformatics analysis begins with raw reads in FASTQ files generated by the sequencer. We use Illumina BaseSpace Sequence Hub to configure and manage many sequencing runs. BaseSpace includes an option to perform automatic adapter removal when sequencing runs are finished. Unless noted otherwise, we include this option in every run. Nevertheless, there are additional tools such as Trimmomatic that could be applied at this step to ensure that all adapter sequences have been removed in FASTQ files.
We then perform a quality control check on each FASTQ file with FastQC to ensure that read quality, read distribution and other QC metrics follow expected patterns.
In the next step, we choose “File-> Import-> From File…” in Geneious to load both the forward read and reverse read FASTQ files. This step displays the “FASTQ Sequences Import” screen:
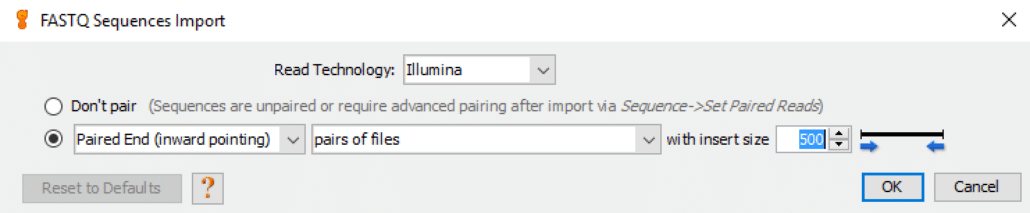
Fig. 2
In almost all cases we choose the following default values:
- Read Technology: Illumina
- Paired-End (inward pointing)
- pairs of files
- insert size: 500
After loading the FASTQ paired-end reads we review the imported sequences.
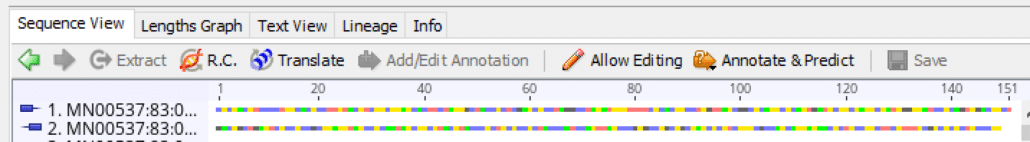
Fig. 3
In Geneious “Sequence View” the forward reads and reverse reads are paired together, record by record, for all reads in the sample. The forward read is shown first with a right-facing box-line icon, and the paired reverse read is shown second with a left-facing box-line icon. The nucleotide bases (A, C, G, T) are color-coded in this example.
We also check the sequence length distribution in the Geneious “Lengths Graph” view:
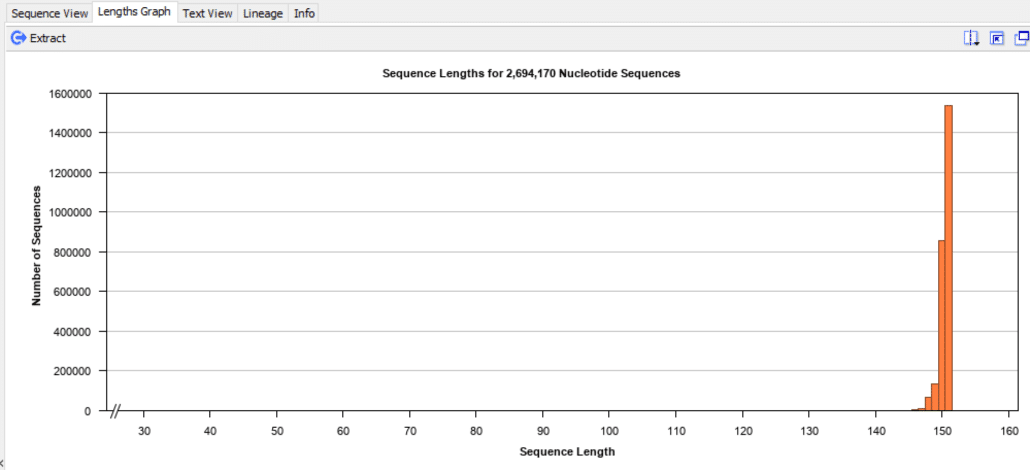
Fig. 4
For this demo run, we used 2×150=300 bp paired-end reads, thus the lengths for the forward reads and reverse reads cluster around 150 bp as expected.
Next, we choose “Sequence-> Merge Paired Reads” which displays the “Merge Paired Reads” screen:
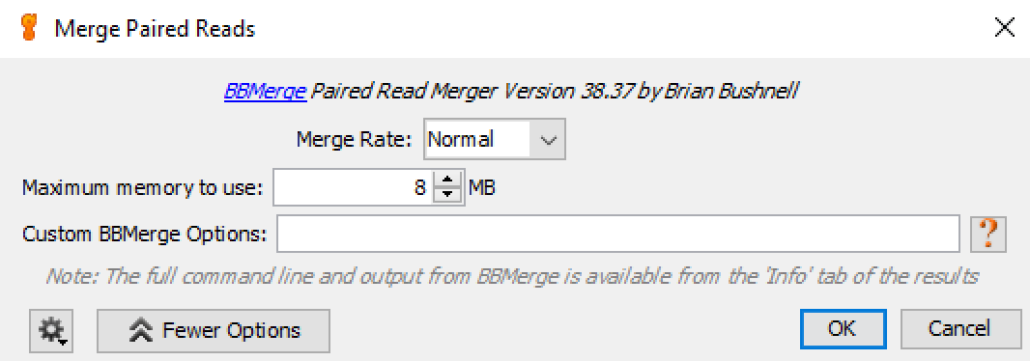
Fig. 5
Geneious uses BBMerge to merge overlapping paired-end reads into single reads. In some cases forward and reverse reads in a paired set of reads have (usually short) overlapping sequences. BBMerge merges these reads to eliminate the overlapping nucleotides, which improves the downstream sequence alignment algorithm. In most runs we choose default options:
- Merge Rate: Normal
- Maximum memory to use: match this option to available RAM
Next, we choose “Annotate & Predict-> Trim using BBDuk” which displays the “Trim using BBDuk” screen:
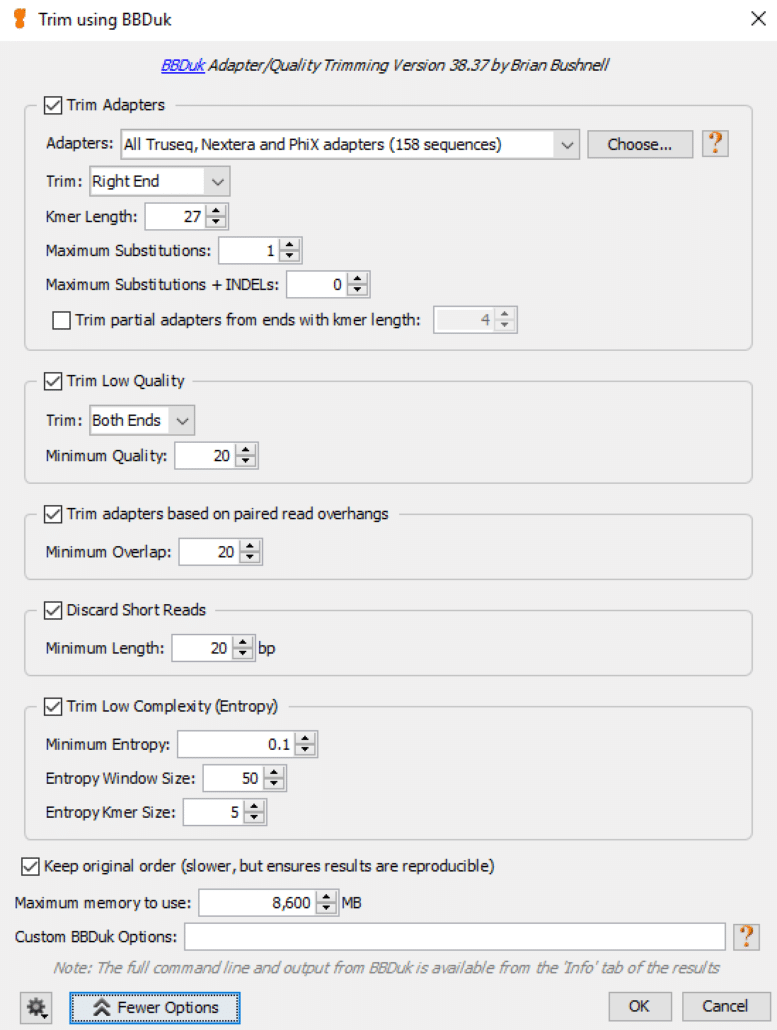
Fig. 6
We usually tweak the default values for this function as follows:
- Trim Adapters: choose defaults as shown in Fig. 6
- Trim Low Quality: set Minimum Quality = 20
- Trim adapters based on paired read overhangs: set Minimum Overlap = 20
- Discard Short Reads: set Minimum Length = 20
- Trim Low Complexity: choose defaults as shown in Fig. 6
- Keep original order: set Maximum memory to use to match available RAM
Next, we choose the function “Align/Assemble-> Map to Reference” which displays the “Map to Reference” screen:
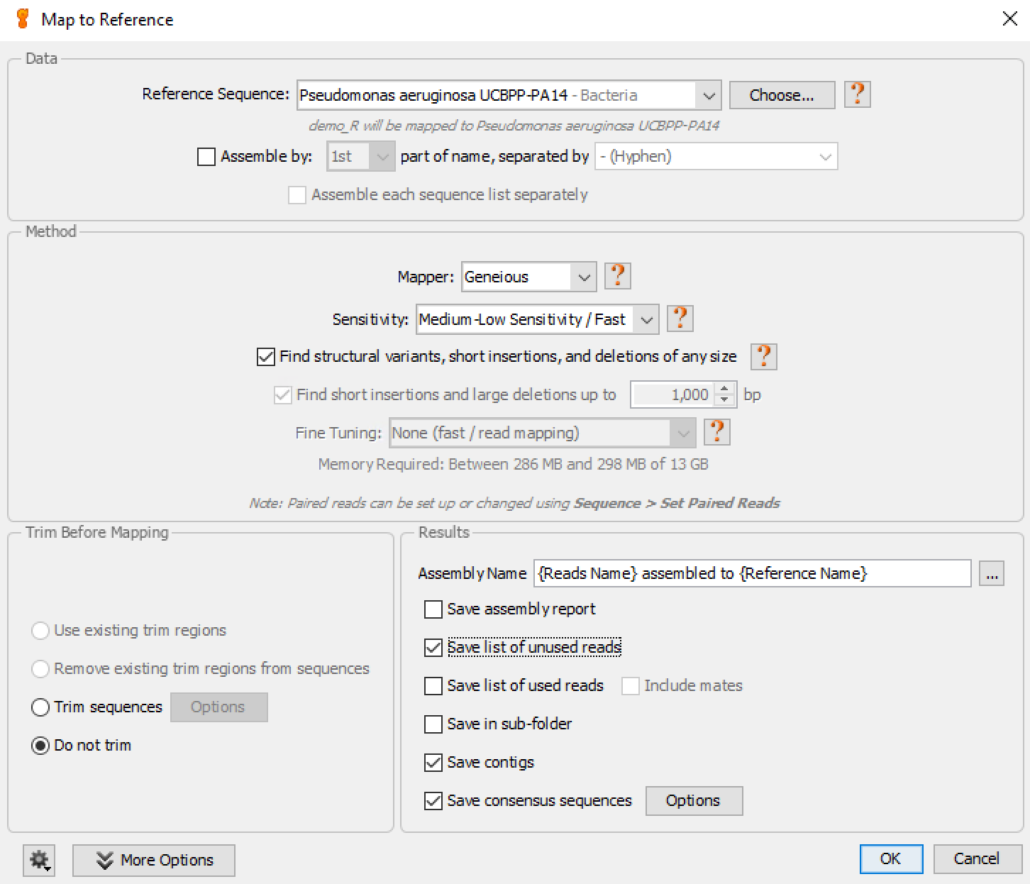
Fig. 7
We usually tweak the default values for this function as follows:
- Reference Sequence: choose appropriate reference genome (this must be installed in Geneious before running the workflow)
- Mapper: choose the default Geneious mapper
- Sensitivity: choose the default Medium-Low Sensitivity/Fast option
- Find structural variants…..: check this option
When the sequence alignment step is finished we choose “Annotate & Predict->Find Variations/SNPs” which displays the “Find Variations/SNPs” screen:
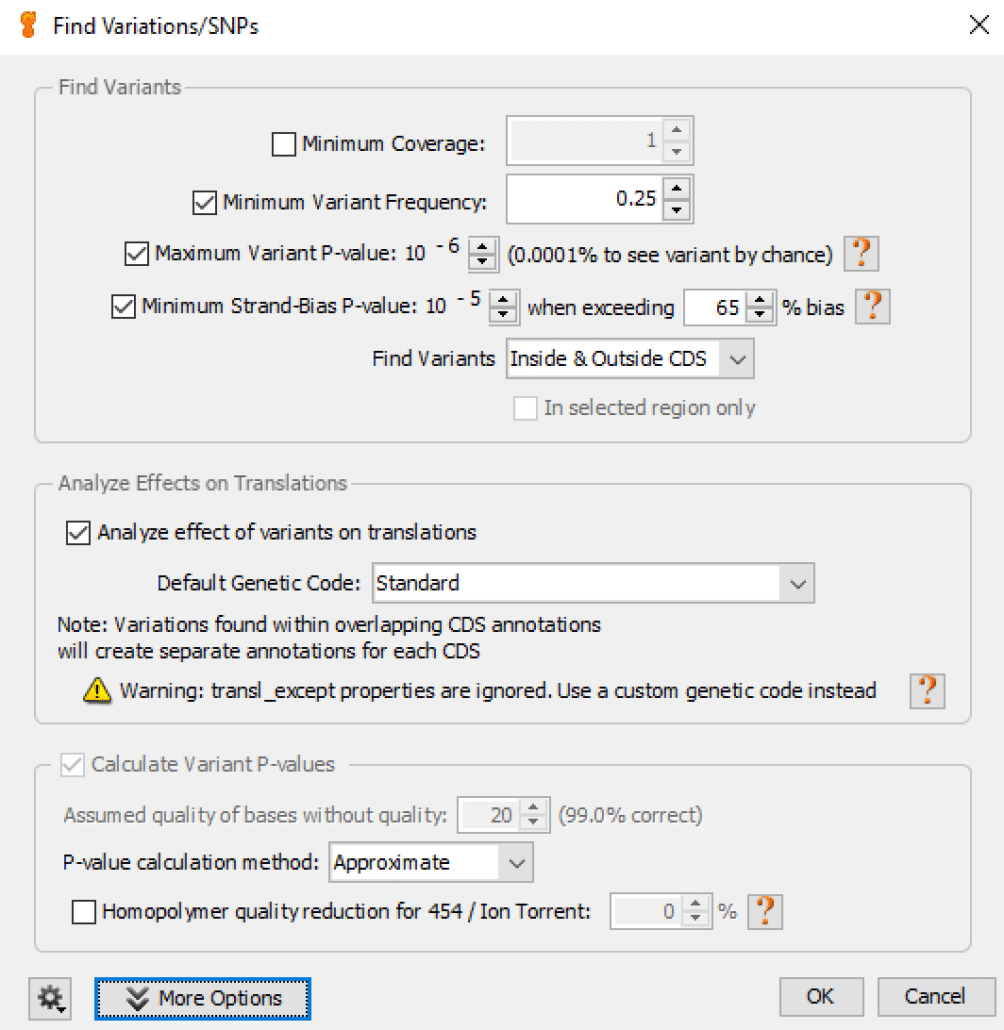
Fig. 8
We usually choose all default values for this function as shown in Fig. 8.
When the variant call function is finished it displays the results in the following format:
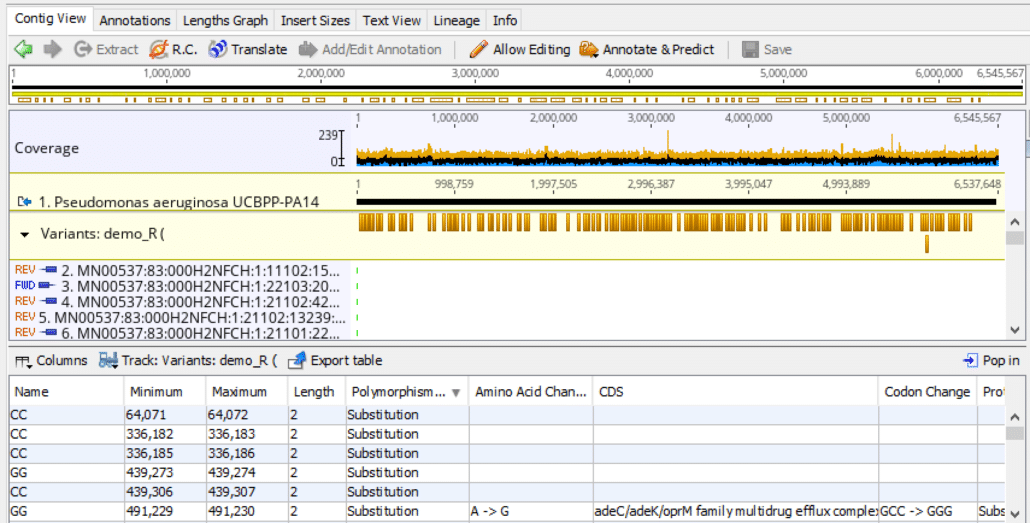
Fig. 9
This basically concludes the Geneious sequence alignment and variant calls workflow. You can check out related articles here:
Sequence alignments with Geneious