Can I use one of my sequenced samples as a reference genome?
We are often asked if clients can submit several samples at once for sequencing and then use one of the samples as a reference genome to compare against the remaining samples. For example, clients may have a single control sample and several treatment samples and would like to use the control as a reference sequence for the treatment samples. In general, this is not feasible on short-read instruments alone like our Illumina MiniSeq, but can be possible using the hybrid assembly of short and long-read sequencing.
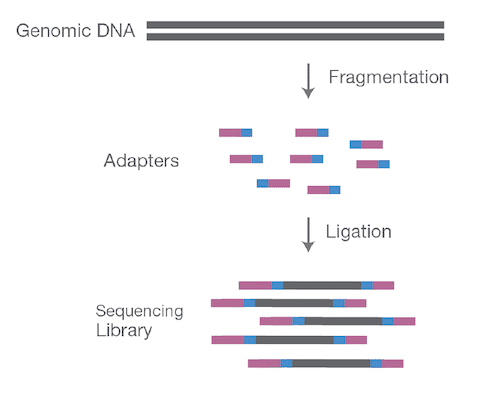
Fig. 1
The Fragmentation Process
During the library preparation process on short-read instruments, intact genomic DNA is (enzymatically or mechanically) sheared into millions of short pieces. As shown in Fig. 1, full-length intact genomic DNA is fragmented into several million short fragments (called “reads”), with each read typically about 150 base pairs (bp) in length. Every sequenced sample is fragmented into millions of reads in this fashion. The fragmented sample libraries are then sequenced on an Illumina short-read sequencer. When sequencing runs are finished, each sample still contains several million 150 bp short-reads.
Aligning to a Reference Genome
Our clients then run sequence alignment and variant calling algorithms of each sample against a known reference genome. They often use Geneious alignment algorithms for this step but many other alignment programs are available. Known reference genomes are often derived from NCBI (National Center for Biotechnology Information) or various other sources. Reference genomes from NCBI and other sites include intact, fully assembled, fully annotated, and validated genomic sequences, which are appropriate for performing sequence alignments and variant calls. For example, a typical fully assembled bacterial genome consists of a single 5 Mb (megabase) sequence. If they align one sequenced sample against an assembled reference genome, they are effectively aligning several million short-reads against a single, intact, fully assembled genome. Sequence alignment algorithms are designed to do this.
Aligning Against Mullions of Reads
In contrast, one generally cannot substitute fragmented sequenced samples for intact reference genomes. If one attempts to align a sequenced sample against another, they are in effect aligning several million short-reads from one sample against several million short-reads from another sample, which yields a very large, exponential number of very short alignments. Basically, the result is a chaotic mess. Thus, in our control-treatment scenario, one cannot arbitrarily choose one sequenced sample as a control and align the remaining treatment samples to it, as they would be aligning millions of reads in each treatment sample against millions of reads in the control sample. Therefore, it is usually recommended that sequenced samples be aligned to known reference genomes as deposited in standard databases like NCBI.
The De Novo Assembly and Hybrid Assembly Solutions
There are alternate solutions to this issue, such as de novo assembly and short-read/long-read hybrid assemblies, but unfortunately, it is not possible to de novo assemble genomes to fully-finished form using only Illumina short-read instruments. However, it’s often possible to de novo assemble to the scaffold level, which typically generates a few hundred scaffolds, i.e. partial chromosomes or genomes (in bacteria, a single circular chromosome equals a complete genome).
In order to get a full genome, it’s generally necessary to use both short-read and long-read instruments and perform a hybrid assembly with those two results. Then, gap filling, scaffolding, and reference genome assembly tools are typically needed in conjunction with manual annotation to complete the genome. We offer long-read sequencing services for hybrid assembly. The resulting complete genome can then be used as a reference genome.