Cost Savings with Multiplex Samples
Multiplex sequencing, or multiplexing for short, is a method of combining two or more samples (pooling samples) in a single sequencing run. Multiplexing can increase the throughput of sequencing runs and save money at the same time.
Conceptually, multiplexing is a fairly straightforward process.
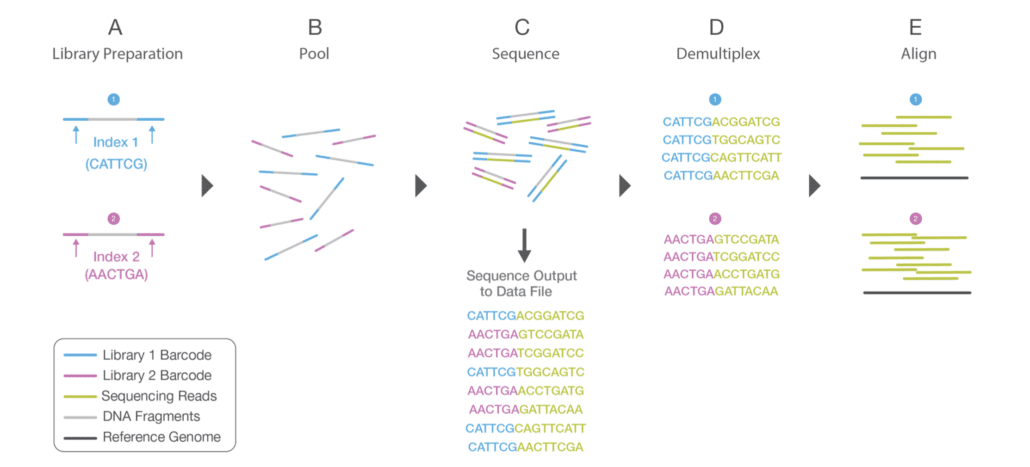
Fig. 1 Multiplex sequencing
Fig. 1 shows how (two) samples are multiplexed and demultiplexed in a sequencing run. During library prep (A), “Index 1” (blue) is annealed to “Sample 1” reads (DNA fragments) and “Index 2” (purple) is annealed to “Sample 2” reads. Each index itself has a unique DNA sequence (six nucleotide bases in this example). The indexed reads are pooled together (B). Note that even though all samples are pooled as one, each sample retains its unique identify via its assigned index. The pooled samples are sequenced on an Illumina sequencer (C).
When the run is finished, a demultiplexing algorithm automatically bins the sequenced reads according to their assigned index (D). All “Index 1” reads go to one bin and all “Index 2” reads go to another bin. There is no user intervention at this step; the demultiplexer runs automatically to properly sort all of the sequenced reads. Each set of indexed reads can then be processed by downstream applications, such as sequence alignment or de novo assembly algorithms (E).
The terms “index” and “barcode” are often used interchangeably. In Illumina-speak we generally use the term “index”, whereas other vendors often use the term “barcode” (hence, the mixing of terms seen in Fig. 1). “Barcode” is somewhat more descriptive in that it implies “unique identifier”, which is the exact function of Illumina indexes.
Although in theory we could multiplex a very large number of samples in a single run, there are practical limits to the degree of multiplexing for any given set of samples. In particular, there is a tradeoff between cost and level of coverage. As described here, the average sequencing coverage can range from 1X (for various reasons) to several thousand, depending on the nature of the sequencing project. If cost is no issue (unlikely), then we can run a single sample that yields very high coverage, but at a prohibitively high cost. Conversely, if cost is an issue (likely), then we can adjust the number of multiplexed samples to yield acceptable levels of coverage at reasonable cost. The latter state is the most common scenario. However, at the other extreme, if we multiplex too many samples in one run, coverage drops to unacceptably low levels, even though the cost per sample is appealing. Therefore, we use cost-coverage calculators and a great deal of experience with assays and sample types to determine the optimal level of multiplexing for any particular sequencing run.
The Cost of Multiplexing Samples
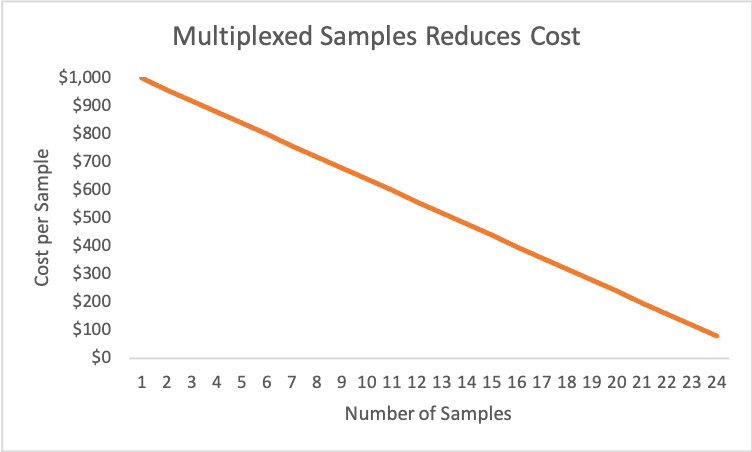
We generally address cost issues on a “Cost per Sample” basis. This is often the most requested unit-of-account for determining sequencing costs.
Fig. 2 The cost of multiplexing samples
Fig. 2 shows a hypothetical example of how multiplexing samples reduces cost. The actual sequencing costs and number of samples that can be multiplexed per run vary considerably. Both variables depend on numerous factors including the sequencing assay, sample type, the level of desired coverage, species-specific genome size, level-of-effort required for library preparation protocols, etc.
The most expensive option is running a single sample, i.e. no multiplexing at all. We rarely make single sample sequencing runs, primarily due to the excessive cost of such runs. The cost per sample declines significantly as more samples are run simultaneously, i.e. increased multiplexing. There is an upper limit to multiplexing that depends on coverage and the limits imposed by pooling, normalization and indexing of samples in specific library prep kits. If too many samples are multiplexed in a single run, the level of coverage may decline to a point where the quality of downstream bioinformatics data analysis suffers, yielding unacceptable sequence alignments, de novo assemblies and other issues.
Although our hypothetical example shows a linear relationship between cost and number of samples, in reality the multiplexing curve may be non-linear. For some assays and sample types even low levels of multiplexing, i.e. running a handful of samples instead of one, can yield significant savings on a per sample basis. The cost per sample can drop by one-half or more by simply running a few, or several, samples in a single run. For this reason, we attempt to multiplex every sequencing run if possible.








Leave a Reply
Want to join the discussion?Feel free to contribute!